All published articles of this journal are available on ScienceDirect.

The Impact of Opioid Analgesics and the Pharmacogenomics of ABCB1 in Opioid Dependence and Pharmacotherapies: A Short Review
Abstract
Pain is extremely multifaceted in nature, with physical injury being just one of its causes. The most commonly prescribed therapeutic intervention for acute as well as chronic pain (postoperative or otherwise) is Opioids. Opioid analgesics act via the opioid receptors (G protein coupled), that are widely distributed in the central and peripheral nervous system as well as the gastrointestinal tract, producing pain relief. The opioid analgesics are divided into distinct categories depending on the receptor type and their potency. Three major types of receptors mediate analgesic effects, namely, mu (μ), delta (δ) and kappa (κ). These are also further subdivided into different subtypes depending on the duration and onset as rapid-onset, short-acting and long-acting. The neurobiology behind opioid analgesia involves the mesolimbic (midbrain) reward system. There is also increasing scientific evidence that plasma pharmacokinetics, along with the CNS distribution of opioids, is greatly influenced by the P-glycoprotein (P-gp) efflux transporter. The ABCB1 gene is responsible for coding P-gp hence it was postulated that variability in the ABCB1 gene could potentially play an important role in determining why there is interindividual variability towards opioids therapeutic interventions
Hence the primary purpose of this review was to compile and understand the research regarding the mechanisms of opioid actions and the effect of genetic variability (ABCB1 gene), on the P-gp transporters regarding their expression/function while also examining the opioid use, abuse and dependence and possibly coming up with some strategies to use pharmacogenomics as a potential tool for solving these issues
1. INTRODUCTION
Pain is extremely multifaceted in nature, with physical injury being just one of its causes. The development of any diseases is often a multifaceted affair. Scientific research and modern advances in medicine have revealed that the level of pain experienced is a combination of psychological and physiological factors. It is of vital importance that the patients experience pain relief, especially after surgical procedures. Adequate pain relief is often deemed to be an important measurement of patient satisfaction and it also helps prevent any post-surgical chronic pain. The most commonly prescribed therapeutic intervention for acute as well as chronic pain (postoperative or otherwise) is Opioids.
Opioids are naturally occurring but are also synthesized commercially with equianalgesic dosage, which allows seamless conversion between agents and different routes of administration. The use of opiates as an analgesic has been long recorded in history, but along with its useful properties, it was also abused as a euphoric agent. To counter the illicit trafficking, use, and abuse, the government has set up numerous narcotic policing countermeasures. Numerous treatment strategies have also been put in place, which help addicts combat their addiction. Presently, substitution therapies for long term maintenance administer methadone and buprenorphine. This substitution therapy has been proven to be the most cost-effective intervention strategy for opioid dependence. However, their rate of success is still not at the optimal level and this could very well be attributed to the limited therapeutic indices when it comes to substitution therapies along with variations in individual responses to therapeutic drugs. Hence, the attrition rate from these intervention therapies remains critically high. Therefore, it is of utmost importance that other underlying factors be explored in depth to arrive at an optimized therapeutic intervention. Though there are many possibilities, this review article will be closely reviewing the P-glycoprotein efflux transporter, which influences intestinal absorption rates, along with tissue distribution and its elimination. A large number of substrates, including opioids, have been known to be influenced by the P-glycoprotein. The other possibility of future therapeutic intervention is the ABCB1 gene, that encodes P-gp, and given its polymorphic variations, it proves to be an ideal candidate to review and study.
2. OPIOID PHARMACOLOGY AND MECHANISM OF ACTION
2.1. Opioid Pharmacology
Since the Sumerian civilization, opium has been cultivated, which dates to 4500 B.C. In the Sumerian cultivation, the poppy plan was known as Hul Gil (the joy plant), There are also Egyptian papyrus records for using opium as a pain relief agent, so since very long-time, opioids have been cultivated, used and abused. The term opioid encompasses all compounds that are produced naturally, synthetically or semi-synthetically, and have morphine-like activity. Morphine is a naturally occurring opioid extracted from Papaver Somniferum (poppy plant). The original opioids consist of alkaloids. There are also a large variety of partial and fully synthetic opioids available on the market. Today the classification of opioids is based on their receptor effect and receptor type (Fig. 1). There are three types of receptor effects, Agonist (e.g. morphine), Antagonist (e.g. naloxone), and Partial Agonist (e.g. buprenorphine). The classification according to the receptor type is four-fold, Mu, Delta, Kappa, and Nociceptin. The last one is the more recently discovered type hence extensive research is required to fully understand its role in the opioid system [1].
2.2. G-protein-coupled Mechanism
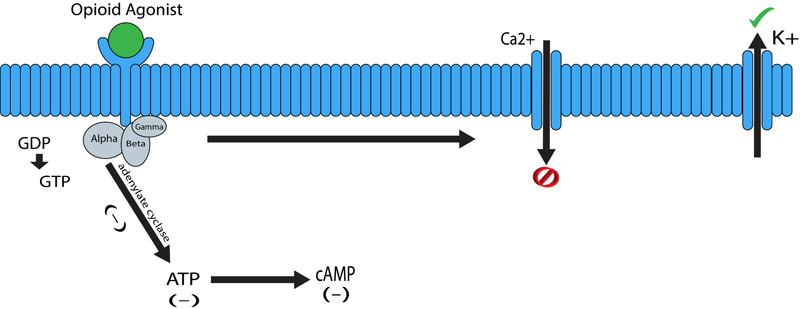
The pharmacological effect of opioids is mediated via opioid receptors, which are the G-protein coupled receptors located both pre and postsynaptically. These act by directly inhibiting the cell signaling by reducing the excitability as well as neurotransmitter release. There is a really good distribution of the G-protein-coupled receptors along with the Central Nervous System (CNS), while fading along the periphery. The Periaqueductal Grey (PAG), locus ceruleus and the rostral ventral medulla all have been known to have high concentrations of opioid receptors. Substantial gelatinosa has a good concentration of opioid receptors present as well [2]. G-protein-coupled receptors are characterized to be heavily involved in antinociception, drug reward, and sedation [3]. G protein coupled receptors are made up of two complexes α-GTP and βγ complex (Fig. 2). Binding of an agonist to the α subunit of the G-protein-coupled receptor leads to its activation thereby causing an intracellular exchange of Guanosine Diphosphate (GDP) molecule with the Guanosine Triphosphate (GTP). The GDP to GTP exchange causes separation of the α-GTP complex from the βγ complex, thus allowing free interaction with other proteins. The α-GTP complex interacts with adenylate cyclase, which in turn causes a reduction in the intracellular levels of the cAMP (cyclic adenosine monophosphate) levels. Cyclic adenosine monophosphate is a secondary messenger required in a lot of biological process, including intracellular signal transduction [4]. These complexes also interact with several ion channels, causing an increase in the activation of potassium channels and inhibiting calcium exchange. The overall effect causing reduced cAMP levels and a hyperpolarized cell (Fig. 2). In the case of neuronal cells this means reduced neurotransmitter release and relay of information [2].

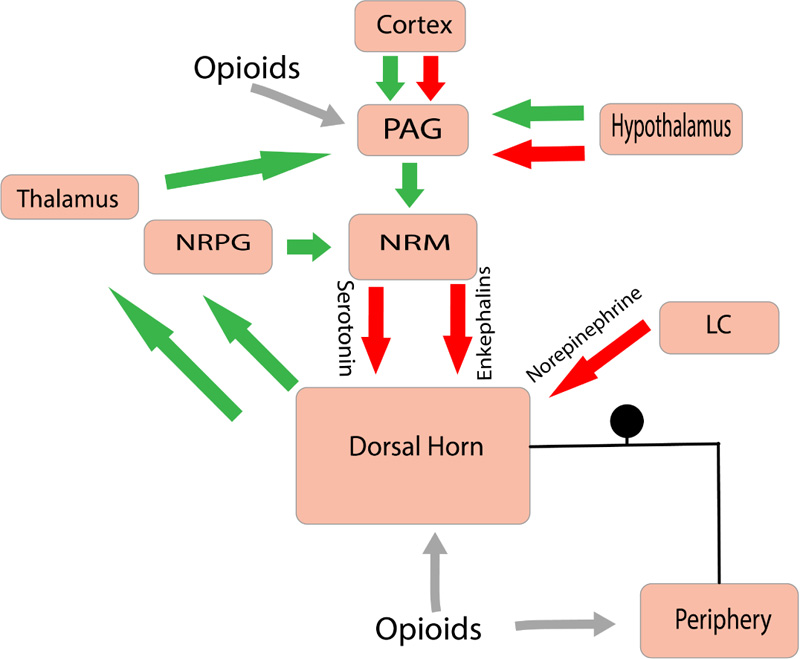
Within the CNS, Mu Opioid receptor (MOP) activation is postulated to be the major analgesia inducing mechanism [5]. The main mechanism of action by the MOP agonists consists of activating the descending inhibitory pathway involving the PAG and the Nucleus Reticularis Paragigantocellularis (NRPG). The overall effect causing an increase in the neuronal traffic through the NRM (nucleus raphe magnus). This causes the increasing activity of 5-hydroxytryptamine and enkephalin-containing neurons that innervate the substantia gelatinosa of the dorsal horn. This inhibitory effect causes a reduction of nociceptive information from the peripheral receptors to the thalamus (Fig. 3). Opioids (Exogenous & Endogenous) have been observed to inhibit the transmission of nociceptive information by directly interacting with receptors present within the dorsal horn (substantia gelatinosa) and the peripheral afferent neurons [6].
Opioids receptors present in the brain are responsible for analgesia, euphoria, sedation and respiratory depression. There are also pre-synaptic opioid receptors on the primary afferents located in the dorsal horn that are also responsible for mediating analgesia [7]. There is also evidence that opioid receptors within the intestine (enteric) may be responsible for influencing gut motility [8]. The opioid receptors are further divided into mu, delta and kappa subtypes. Among them, mu opioid receptors are the best characterized and are still used as the primary target for opioid therapeutic interventions [9]. These are also further subdivided into different subtypes depending on duration and onset as rapid-onset, short-acting and long-acting. For the purposes of this review paper, the mu-opioid receptor agonists will be reviewed as they exhibit all the common opioid effects such as analgesia and euphoria along with its side effects such as constipation, nausea and respiratory depression.
3. ANALGESIC MECHANISM AND THE COST OF ADDICTION
3.1. Opioid Analgesia and Side Effects
Research has established that acute administration of drugs of abuse like opioids leads to an increase in the extracellular levels of dopamine in the nucleus accumbens, this leads to the activation of the limbic system, which is primarily involved with emotion, motivation, and reward. This has been demonstrated using heroin, morphine and methadone [10]. The pre-synaptic receptors on the GABA neurons usually inhibit the dopaminergic neurons in the VTA (Ventral Tegmental Area). These dopaminergic neurons extend into the reward centers of the brain from the VTA. When there is mu-opioid activation by inhibiting GABA neurons, the dopaminergic neurons release copious amounts of dopamine into areas such as the Nucleus Accumbens (NAc) leading to a sensation of euphoria and reward. Along with the NAc, the involvement of the lateral hypothalamus, hippocampus and Periaqueductal Grey Area (PAG) have also been implicated in the opioid reward cycle [10]. Endogenous opioid peptides and the endocannabinoid system are postulated to be involved in this reward effect of opioids [10]. Experiments have also indicated that glial cells may also be involved in modulating the opioid reward response [11]. Hence multiple pathways working in unison are deemed to be responsible for the opioid analgesic effect.
Along with desired effects, there are numerous adverse effects that are commonly present with opioid use. The most common is constipation and occurs in around 40-95% of people being administered the drug [12, 13]. Other adverse effects are nausea, vomiting, and respiratory depression, which can result in hypoxia and hypercapnia. It has been postulated that the opioid receptors located in the respiratory control centers in the brain stem might be responsible for this [14]. Although very rare, death resulting from opioid overdose can also happen in a clinical environment [15].
3.2. Opioid Addiction and Cost
Any addiction follows the same steps beginning with tolerance, followed by, withdrawal; continued usage of the drug for long amounts of time, unsuccessful attempt at reducing the use, replacing all social, occupational and recreational activities with the drug and continue usage despite being aware of the harmful consequences. Tolerance is a very common issue arising in almost all opioid treatments and is the result of losing drug potency over time, which results in increased dosage requirements to maintain the effect. Two factors that affect this are pharmacokinetic and pharmacodynamic tolerance [16]. Pharmacokinetic opioid tolerance is defined as the upregulation of drug metabolism or its transport due to repeated opioid administration. The main causes of this reaction are decreased bioavailability of the drug along with increased clearance. These changes, in turn, decrease the exposure of the CNS to the drug, leading to an increasing demand. Pharmacodynamic tolerance relates to changes in the receptor availability and its activation along with changes in the descending pathway that results in decreased drug efficacy. The proposed mechanisms of this tolerance are alterations in receptor density, recycling and desensitization [17].
Another reason pertaining to addiction is over prescription of opioids, while avoiding the use of alternative interventions have led to an opioid epidemic in the USA, the total annual cost out of which has been estimated to be around $55.7 billion [18]. Approximately 30% of people living in the United States suffer from some form of chronic and/or acute pain, with more than 40% of the adult population experiencing chronic pain. A high number of patients suffering from chronic pain are directly responsible for the increased opioid prescriptions as a viable therapeutic intervention [19]. It has been estimated that in the year 2014 alone, around 245 million opioids were prescribed, 65% of which were used for short-term therapeutic interventions [19]. Even though opioids provide rapid relief from nociceptive signals, their effectiveness in the treatment of chronic pain is still unclear. According to the CDC a 10-day opioid treatment can also lead to addiction; hence opioid use for acute pain is also heavily associated with increased risk of long-term opioid use [20].
3.3. Need for New Therapeutic Interventions
The use of opioids for pain management has significantly increased in the past two decades leading the increased deaths due to over dosage. Hence there is a significant need for therapeutic interventions that balance treating pain, while minimizing risks for opioid abuse. A validated screening technique that predicts risk and identifies potential problems could very well help in selecting patients for opioid therapy. The present screening strategy for potential opioid abuse includes the assessment of premorbid and comorbid substance abuse; assessment of aberrant drug-related behaviors; risk factor stratification; and utilization of opioid assessment screening tools and is deemed ineffective at present. A novel screening technique relies on pharmacogenomics to determine the possible reaction of the patient. It has been observed from previous research that genetic predisposition plays a key role in how the body processes/reacts to the any drug [21]. The current the “trial-and-error” approach to prescribing medicine is costly and causes delays in effective care with an increased potential of drug abuse leading to overdose as well.
4. VARIABILITY IN DRUG RESPONSE
Since the inception of pharmacology, it has been documented that individuals have had varied responses to administered drugs and these variations are either inherited or acquired but are always present. This is one of the factors that makes optimizing a dosage regimen a real challenge as most drugs only effective in 25-60% of patients [21]. This results in patients not being able to respond to the first recommended therapeutic intervention fully. For example, nearly 38% of depression patients, 40% of asthma patients, 43% of diabetics, 50% of arthritic and 75% of cancer patients exhibit zero response to the first recommended therapeutic intervention [22]. Variability is not only seen towards the same drug and but also towards the dosage quantity. What has been optimized to be the most effective dosage might prove to be lethal to someone else. A continuous and vigil drug monitoring is strongly recommended for drugs that are known to induce serious side effects, mainly to avoid any unexpected outcomes [23]. This situation can worsen if the recovering patients administer some other drug for some other existing condition or if he/she feels that the current drug is not working. There could be a harmful drug-drug interaction that could undermine the treatment in a bad way [23]. A good example of variation is the study pertaining to the drug simvastatin. A daily dose of 40 mg/day of Simvastatin is recommended to combat blood cholesterol levels. In the experimental study involving a total of 156 participants, 95% exhibited low cholesterol, but the remaining 5% did not show any change even at higher dosage of around 160 mg/day. It was later postulated that genetic polymorphisms in the genes coding for ATP-binding cassette sub-family G member 2 (ABCG2) and HMGCR (3-Hydroxy-3-Methylglutaryl-CoA Reductase) gene contributed to these zero effects in some participants [24]. Women, in general, seem to be more addicted than men across all ethnicities [25].
4.1. Factors Affecting Individualized Drug Responses
Why do individuals respond in a specific way can very well be attributed to a variety of complex factors, genetic makeup being the first. Mutations such as SNPs (Single Nucleotide Polymorphisms), deletions, additions, duplications can result in undesired proteins with undesired effects. Physiological factors such as age, gender, body size and mass, ethnicity must be considered along with genetic predispositions. The next set are environmental factors such as diet, smoking, drinking, exposure to harmful toxins and lastly pathological factors such as liver, renal functionality, diabetes and/or obesity also contribute to the individualized drug responses [26]. According to the hypothesis of [27], genetic factors alone make up for 20-95% of the variability an individual will have towards a drug. It is worth nothing that of all the aforementioned factors, genetic factors are the only one that remain permanent [28].
4.2. Varying Individual Drug Response Determinants
Observing signs and symptoms exhibited by the patient (often heavily influenced by familial history) helps to successfully diagnose several diseases. The disease manifests itself in the patient, the patient exhibits the appropriate symptoms, then and only then the clinicians attempt to tackle it. This has been the normal protocol since the beginning of medicine. At times this proves to be an outdated methodology as some diseases don’t exhibit symptoms till the final stage. A novel way around this is examining genes and their variants associated with specific disease conditions, and these altered genes are also responsible for the varying individual drug. The human genome project’s contribution to this endeavor has been significant.
Pharmacogenomics, which is an amalgamation of principles from both pharmacology and genetics enables scientists to pinpoint the particular genetic loci responsible for the varied drug response [29]. Scanning the entire genome for SNP variability, analyzing multiple genes at the same time, haplotype analysis and better understanding the expression of various genes could very well lead to better therapeutic interventions [30]. Genetic polymorphism can heavily alter with the pharmacokinetics as well as the pharmacodynamics of the drug. Pharmacokinetics deals with how the drug interacts with the body and how much is actually required for producing an optimal effect while pharmacodynamics deals with how the body reacts to the drug, which primarily involves the way receptors, ionic channels and other enzymes respond to the drug [31]. Genetic variations can also target membrane proteins involved in the transportation of drug molecules as well as the enzymes that help metabolize drugs. Variations at a single base on the genome i.e. SNPs (Single Nucleotide Polymorphisms) or a group of highly similar SNPs (haplotypes) can affect the overall response of the drug [31] (Fig. 4).
Changes in protein structure and expression resulting from mutations in the coding region affects the quality as well as quantity of transcribed protein. Enzymatic mutations directly affect functional protein properties and protein-protein kinetic rate constants, resulting in varied drug responses. As aforementioned that among all the factors affecting varied drug response, genetic factors are the only ones that remain permanent [32], hence for the purposes of this MAP, the ABCB1 gene and P-glycoprotein efflux transporter (coded by ABCB1) and their involvement in opioid response were focused.
5. P-GLYCOPROTEIN
5.1. P-glycoprotein Efflux Transporter
Among the human efflux transporters, P-glycoprotein (P-gp), encoded by the ABCB1 gene, has been postulated to be responsible for producing the variability related to opioid pharmacokinetics [33]. The interest in P-gp came about when pharmacologists observed an overexpressed glycoprotein that was isolated from a multi-drug resistant tumor cell [33]. The role of P-gp in cancer cells and multidrugg resistance has been extensively studied with special attention to resistances towards chemotherapeutic drugs, however, in the past decade, its involvement in the pharmacokinetics of opioids has also been studied due to its expression in normal tissues [34].
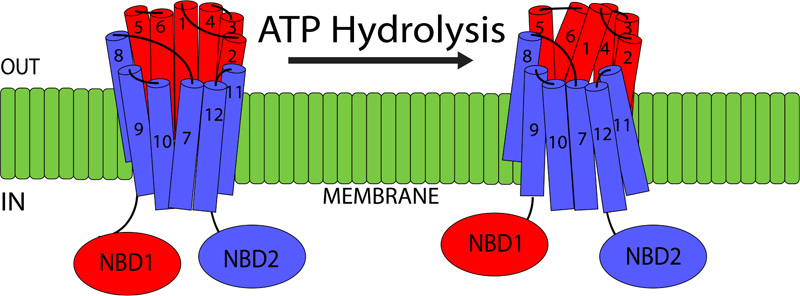
5.2. P-glycoprotein Structure
P-gp is part of the ATP-binding cassette transporter superfamily, transporting the substrates across the membrane in an ATP-dependent manner. This 170 kDa transporter is present in the lipid bilayer and is composed of two homologous regions. Each region is made up of six Transmembrane Domains (TMDs) and one intracellular NBD (Nucleotide Binding Domain) [35] (Fig. 5). NBDs are important as they are responsible for utilizing ATP during the active transport process. There is also a flexible linker region present between the two homologous halves, that not only helps with the interaction between the two regions but also facilitates communication between the two NBDs [35]. The TMDs are postulated to be the main sites where substrate recognition occurs. Recent research has observed the following TMD sites, TMD 5, TMD 6 as well as TMD 11 and TMD 12 as the major substrate-binding regions [36]. However, further research is required to accurately predict the exact mechanisms by which P-gp recognized its substrates and transports them. There is a significant difference in the size and structure of the substrates, that are transported by the P-gp. These range from small organic cations, amino acids, carbohydrates to large molecules such as proteins and polysaccharides. Currently, there are over 480 identified substrates that are transported by P-gp [37]. Numerous P-gp inducers as well as inhibitors have been identified as well. The inhibitors act via competitive binding and transport, while research has also shown a modification of allosteric binding sites affecting P-gp activity [38].
5.3. Expression, Location and Function
P-gp, an efflux transporter, is expressed in various human tissues and is responsible for transporting substrates out of the cell into the extracellular space. It is expressed on the apical membranes of epithelial cells present in the stomach, small intestine, renal proximal tube and the colon. It plays a protective function by reducing substrate absorption and increasing its elimination [39, 40]. P-gp is also known to play an important role in drug distribution into organs such as placenta, testes, pancreas, and the Central Nervous System (CNS) via the Blood Brain Barrier (BBB), by preventing substrate accumulation [40]. P-gp is also highly expressed in the liver hepatocytefig. It has also been shown to have similar substrate specificity as well as colocalization with CYP3A4 metabolizing enzyme. This could be indicative of a coordinated function when it comes to the elimination of foreign substances (xenobiotics) thereby protecting the tissue [41]. Thus, the most opioid relevant P-gp sites are those influencing intestinal absorption, distribution within the CNS via the BBB and helping with its elimination. As aforementioned P-gp expressed on the luminal membranes of the intestinal epithelial cells transports in a basal to an apical direction, while forcing out substrates from the enterocytes into the gut. This is the mechanism that is utilized to maintain/limit the absorption and bioavailability or orally administered substrates [41].
In the brain, the location of the P-gp is within the apical membrane of the brain capillary endothelial cells that form the blood brain barrier and just as in the intestinal tissue, the substrate transport is from basal to apical direction. There are a lot of aspects that contribute to determining the transport of substrates across the BBB. There are tight junctions present between the capillary endothelial cells, as a result, any chance of passive transport across the epithelial surface is negated. As a result, the drugs need to pass through the cells to reach the extracellular space within the brain. The concentration gradient between cerebral blood and the extracellular fluid within the brain is small as compared to the gradient between the intestinal lumen and mucosal blood. The cerebral blood flow is quite rapid compared to the bulk flow of cerebral fluid, meaning that the extruded drug is rapidly removed from the blood brain barrier and is not reabsorbed [42]. Studies in mice have demonstrated that P-gp knockout mice have 17-83-fold increase in the P-gp substrates within the brain as compared to the 2-3-fold increase in the plasma, live, kidney and small intestine [43]. In human studies, the administration of cyclosporine (P-gp inhibitor) exhibited a 87% increase in P-gp substrate in the BBB, whereas there was only 6-17% increase in the plasma [44]. Hence there is good scientific evidence behind the ability of P-gp in influencing the CNS entry of its substrates.
Hepatic P-gp is located on the apical membrane of the hepatocytes and functions in unison with the basolateral influx as well as the canalicular efflux to transport its substrates from the blood into the bile. Animal experiments have demonstrated that in P-gp knockout animals, the biliary excretion of doxorubicin decreased by 62-84% [45]. In human studies, the administration of inhibitors caused 42% decrease in excretion of digoxin [46].
When it comes to renal elimination, P-gp is expressed on the apical, membrane of the renal proximal tubule of the kidneys. Thus, the P-gp substrates undergo tubular secretion and hence have an elimination rate that is higher than the glomerular filtration. Thus P-gp can influence the rate of clearance, mainly by facilitating their kidney excretion. However, how does the renal clearance determine the overall clearance, as well as the plasma concentration, is still unclear. The research till date has shown that P-gp has a minor impact on excreting drugs from the cells as compared to its action of inhibiting drug uptake [47].
In conclusion, thus, it is evident that P-gp can limit intestinal absorption as well as drug distribution within the body and it can also facilitate the renal and biliary excretion. However, when considering opioids, the main site of action is the BBB, where it can limit the passage of its substrates and mediate the drug access to its site of action in the CNS.
5.4. P-glycoprotein & Opioid Transport
Research pertaining to structure-activity relationship analyses have revealed different chemical moieties that are deemed important for the P-gp transport of various neurotransmitters, potassium channel modulators, vasodilators, anti-inflammatory drugs, and anti-cancer drugs. However, many of these tests were not performed for the opioid class of drugs and hence the interactions opioids have with P-gp are difficult to predict based on the chemical structure-activity alone. Hence, the analyses concerned with P-gp have relied on in vitro transport as well as ATPase activity and animal in vitro transport studies.
5.5. In vitro and Animal Studies
In vitro as well as In vivo experiments have identified numerous exogenous as well as endogenous opioids that are P-gp substrates. Usually, there have been no disagreements pertaining to the substrates however, there have been some discrepancies with reference to the investigative methods. From previous research, it was observed that the most reliable method from in vivo studies was from gene knockout studies, that have a lot of advantages over in vitro methods. However, both types of methodologies agree that morphine as well as methadone are transported by P-gp. Experiments involving morphine transport and inhibition in the knockout of P-pg, it was reported that there was 5.2 times increase in the plasma AUC (area under the curve) of oral morphine and also 1.2 times increase in morphine uptake in the brain [48]. It was also observed that there was 1.5 up to 4.5 times increase in concentration in the brain, 3 times increase brain half-life and almost 4.4 times increase in analgesia with no influence on clearance [49]. Likewise, studies with methadone also observed similar results as morphine [50]. Thus, it was clear from animal studies that P-gp affects opioid absorption in the intestine, its distribution in the brain (leading to the antinociceptive effect). There is however, no effect on the elimination. There is also limited evidence with regards to the involvement of P-gp and the distribution of endorphins.
5.6. Elimination
In research experiments till date, quinidine and nelfinavir (P-gp modulators) were used. This difficult to determine the actual contribution of P-gp to the biliary excretion as they cause changes in the activity of hepatic enzymes (involved with morphine and methadone). However, it is possible to estimate the effect of P-gp modulation on renal excretion of morphine and methadone. Till date, no direct relationship between P-gp inhibition and effect on renal clearance has been observed in researches [51]. Research involving rodents and the influence of methadone and morphine on absorption, distribution and elimination, observed significant results, but these were not replicable in in vivo human studies.
5.7. Variability in P-gp Expression and Function
From previous in vitro and the in vivo research, it has been observed that variability in P-gp expression is the main reason behind the difference in euphoria/reward response to illicit opioids, the development of dependence, and possible changes in the endogenous opioid system [52]. These individual differences are also the reason behind the variability in responses to opioid substitution therapies. Hence identifying individual variability in P-gp activity is a crucial factor for understanding as well as predicting risk of opioid dependence as well as their substitution treatment.
Significant variability in P-gp expression has been observed in most of the tissues in which these transporter pumps are present. In the intestine, this expression varies from 2 to 10 folds [52], 3 to 4 times in lymphocytes [53]. Research has also shown a large variability in P-gp expression within brain microvessels [54, 55]. In the liver it was observed that ABCB1 messenger Ribonucleic Acid (mRNA) expression in healthy individuals varied 200-fold with a corresponding 20 to 50 times variability in protein levels [56]. Numerous compounds have been suggested to be responsible for producing this variability in the P-gp expression, the majority of which are its own substrates. There are P-gp inducers that are known to act at the transcriptional level, increasing ABCB1 mRNA expression [57, 58]. Drug-Drug interactions (methadone and P-gp inducers) have also been attributed to opioid withdrawal in MMT (Methadone Maintenance Treatment) patients [59]. Other opioids such as oxycodone has been shown to increase the expression of P-gp in kidneys, liver, intestine and brains of rats and this increase ranged from 1.3 to 2 times [60]. Animal studies involving repeated morphine exposure have been associated with P-gp expression as well [61]. Compared to animal studies, less studies have been conducted in vivo in humans to study the effect of proposed P-gp inducers and opioid distribution, hence further research is required to determine the long-term effects of opioid administration on treatment response. Along with pharmacological effects, there are other stressors such as cell damage/stress, cytotoxins, X-rays are also possible P-gp inducers [36]. In another experiment, peripheral inflammation in rats resulted in a significant increase in P-gp expression within the BBB while decreasing morphine brain uptake as well [62].
Numerous medications inhibit P-gp transport, these either high affinity substrates and bind non-competitively, thereby preventing binding of other substrates. They are also efficient inhibitors, targeting the ATP hydrolysis, which is the main component required for transport [63]. Other dietary compounds such as flavonoids have also been exhibited as potential P-gp inhibitors and some excipients found in drug preparations have also been shown to inhibit P-gp function [64].
ATP-Binding Cassette (ABC) transporters. The ATP mediated transport of solutes across the membranes against a concentration gradient, is primarily mediated by the family of proteins known as the ATP-Binding Cassette (ABC) transporters [65]. The presence of these transporters in not only all eukaryotic species but also in bacteria and archaea, assesses its evolutionary and scientific importance [66]. The first of the nine ABC drug transporters ABC (MRPs or MRP1) in humans was cloned in the year 1992 [67]. ABCC/MRP1 is encoded by the ABCC1 gene on chromosome 16p13.1. Since 1992, MRPs, or MRP like proteins have been identified in almost all eukaryotes, however, ABCC proteins have not been observed in any bacteria or archaea [67].
In humans, the ABC superfamily of proteins is composed of 48 genes and 3 pseudogenes [68]. These 48 ABC transporters are further classified into 7 subfamilies, depending on the relative degree of sequence homology. This subclassification ranges from A to G [68]. The ABC subfamily is composed of 12 proteins and it has been observed that 9 of these proteins are responsible for mediating the ATP-dependent transmembrane efflux transport against their concentration gradients [69, 70]. In bacteria, ABC transporters are also grouped as exporters (responsible for exporting lipids, drugs, sterols, primary as well as secondary metabolites) and importers (responsible for taking up nutrients, trace materials, vitamins and biosynthetic precursors) where as in eukaryotes ABC efflux pumps are mainly exporters except for ABCA4 [71].
6. ABCB1 GENE
6.1. ABCB1 Genetic Variability and Structure
The ABCB1 also known as Multidrug Resistance 1 (MDR1) encodes P-glycoprotein (P-gp), which is responsible for the efflux of various carcinogens, metabolites and chemotherapeutic agents [72]. ABCB1 is highly polymorphic with variant alleles found in varied frequencies in different populations. The ABC superfamily of proteins is composed of 48 genes and 3 pseudogenes, these 48 ABC transporters are further classified into 7 subfamilies ranging from A to G. As it can be observed mutations in the allele can lead to a variety of functional consequences, affecting ATP binding and hydrolysis, gene expression and loss of transport activity altogether [73].
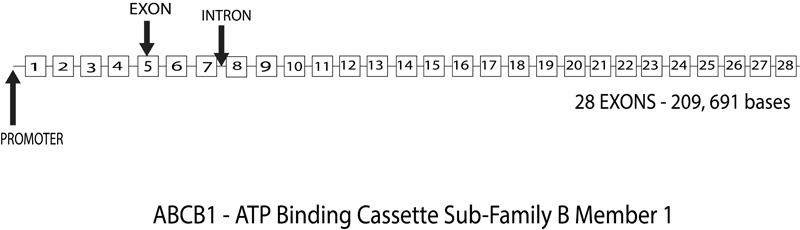
The 120 kilobases (kb) human ABCB1 (ATP-binding cassette transporter B1) gene is located on chromosome 7 at q21.1and consists of 28 exons numbered from -1 to 28 (Fig. 6). Research has postulated two promoter regions, first at the beginning of the exon at -1 and the second at exon 1, which is found to be preferentially expressed in a lot of cells. However, research has also found that the actual translation site starts at 2 and the coding sequence consists of 26 exons, 2 to 28 [74]. Numerous transcription regulatory elements are in and around the first 300 base pairs upstream from the transcription start sites. There is also an enhancer region located at 8kb upstream [74]. Being an efflux transporter, ABCB1 is located on the apical surface of epithelial cells preventing intestinal absorption, protecting the brain as well as the placenta form any xenobiotic material as facilitating renal and biliary excretions as well [75].
6.2. Single Nucleotide Polymorphism (SNPs)
The ABCB1 polymorphisms were identified for the first time in 1989 [76], since then, most of the researches dealing with ABCB1s were based and focused around acquired mutations in tumor cells mainly associated with Multiple Drug Resistance (MDR). It wasn’t until the year 2000 that the first screening for genetic variability pertaining to ABCs was conducted by Hoffmeyer and associates that hinted at the heritable nature of ABCB1s, causing mutations related to drug dispositions [77]. This important study identified 15 naturally occurring SNPs in healthy Caucasians and identified a variant allele of a synonymous SNP, C3435T. This variant was found out to be associated with a decrease in P-gp expression in the duodenum resulting in increased intestinal absorption of digoxin, a P-gp substrate. Because of this landmark study, C3435T SNP became the most widely studied variant of ABCB1. Research till date has observed ABCB1 be highly polymorphic, with over 1200 polymorphisms reported in the gene. There are also over 60 SNPs currently identified within the exonic sequence [78].
6.3. ABCB1 Haplotypes
In the Caucasian population alone, there are 64 discernable haplotypes have been reported. Research has observed significant disequilibrium between the linkages especially between the three variant alleles 1236, 2677 and 3435 [79]. The most common haplotype is termed as MDR1*1 (wild type) and MDR1*2 that have variants at positions 1236, 2677 and 3435. Together these account for around 63% of the chromosomes [79]. There is however a considerable ethnic variability observed among the various SNP and haplotypes of the ABCB1 gene. For example, the frequency of the wild type allele C at 3435 ranges from 84% in African Americans to 34% in the South-West Asian population [79]. On the other hand, the frequency of MDR1*2 haplotype is only around 6-7% in African Americans as compared to 27% observed in Caucasians [80]. It has been observed that allele variants of this gene are ethnic dependent, for example, SNP 3435C > T has a higher frequency of occurrence in Asian populations (60-70%) but operates at a lower frequency (34-42%) in Caucasians. In a study by Sakaeda and colleagues, Patients with mutant allele, 3435C > T were administered a single dose of digoxin, exhibited low serum concentrations of digoxin [81] whereas another study by Verstuyft and colleagues, concluded that higher plasma levels of digoxin [82]. Hence the mechanism of action is not fully understood, and further research is required to acquire concrete proof. Asian population was reported to have high concentrations (up to 56%) of the haplotype 1236C > T/2677G > T/3435C > T [83]. A study by Kimchi-Sarfaty et al., observed that patients with this haplotype had normal transporter properties, although transport inhibition by small modulators was mainly affected [84]. There have been so many contradictory studies in humans, and one of the reasons for this could be that are other polymorphisms that haven’t been identified yet or could be two complex dispositional mechanisms operating at the same time. For example, a widely used immunosuppressant substrate, CYP3A4, used in patients with kidney, heart transplants is also transported by ABCB1 [85]. Similarly, digoxin can be simultaneously transported by OATP and ABCB1 [86]. The African populations have even lower 3435C > T alleles, compared to Asian and Caucasians, this might be one of the reasons why there is a reduced incidence of renal carcinoma in African populations [86]. Such varied genetic differences are the reason behind a push for genetic screening to avoid any potential drug overdose/abuse.
6.4. Functional Consequences of ABCB1 Variability
After the landmark study by Hoffmeyer, in 2000, there was a strong push to study the C3435T SNP and there were a lot of experiments based around digoxin and C3435T [77]. Parallel to this study, there was an increasingly growing need to understand the mechanisms behind the SNP effects, mainly on overexpression and function of P-gp. This need prompted numerous in vitro as well as in vivo experiments. It has been established that ABCB1 SNPs and their haplotypes directly affect the expression and function of P-gp, with clinically relevant effects. The proposed structural model of P-gp provides some clues about where the mutation might occur, and how this might affect the P-gp function. For example, any mutations at the NBDs will affect the ATP hydrolysis necessary for transport. Any mutations in the flexible linker region located between the homologous halves of the P-gp might result in the intonation of the transport capacity. Unfortunately, the three common non-synonymous SNP do not lie within any of these regions, hence predicting what functional consequences might occur due to mutations is challenging. Mutations in the common synonymous SNPs of ABCB1 (C1236T and C3435T), might affect the P-gp expression rather than function, but they might play a role in both. The next few sections will discuss the research conducted in the past two decades, including all in vitro expression analyses and ex vivo human clinical studies mainly to observe and understand the impact of ABCs, particularly ABCB1 genetic variability on P-gp function and expression. There are clear ethnic differences in the frequency of ABCB1 mutations and the functional effects of the SNPs also differ among the ethnicities.
6.5. In vitro Expression and Function
There have been several experiments that used cells transfected with wild type or mutant P-gp to examine the effects on transport and expression. Despite utilizing almost similar experimental techniques, the results have significantly varied depending on the P-gp substrate tested and the expression analyses employed. Now we will look in detail, each of the mentioned SNP:
A61G:
Both the studies investigating the synonymous A61G SNP did not observe any effect on expression however, paclitaxel transport was reduced in one study [84] but the other study did not find the same effect [87].
C1236T:
There were two In vitro studies carried out to examine the functional effects of the C1236T on P-gp. In the first study by Salma and colleagues (2006), variant expressing cells showed a reduced transport capacity for some compounds [88]. While another study showed that the T variant along had no significant effect [89]. Since C1236T SNP causes a change in the mRNA folding, there is no change observed in the stability of the mRNA [90].
C3435T:
This was the first SNP to be identified and has been described as a heritable, clinically relevant ABCB1 mutation. This has been extensively studied in vivo and of the in vitro studies did report decrease in the in vitro transport of compounds. The mechanisms behind its effect on P-gp are still debatable with the latest studies indicating mutations in the secondary structures of the mRNA as the probable cause [90].
G1199A:
In vitro experiments have shown that G119A SNP has no effect on the expression of P-gp, however, its effect pertaining to P-gp functions have been varied. The majority of the studies indicate no effect on P-gp function, its variant did indicate a decrease in the transport of some compounds such as rhodamine and verapamil. They also indicated an increase in the transport of other compounds such as vinblastine, vincristine, amprenavir, indinavir, lopinavir, ritonavir, saquinavir and doxorubicin [91]. So, it can be concluded that presently the functional consequences remain unclear.
G2677T:
The outcome of the experiments involving G2677T SNP has been consistent with a decrease in paclitaxel transport in the variant transfected cells. Studies using other compounds/drugs have also reported a decrease in transport and no significant effect on mRNA folding was also observed [92].
Additionally, there are various experiments investigating the variant haplotypes, namely, 61G/1199A, 61G/2677T and 1199A/2677T. The experiments reported that none of them were directly associated with altered P-gp expression they did all exhibit a decrease in the transport of compounds [90].
6.6. Ex-vivo Expression and Function
The best alternative to in vitro studies has been ex vivo analysis of human tissues. Analyzing the P-gp expression in isolated human tissues has a distinct advantage over in vitro techniques as the human tissues closely resemble the in vivo P-gp expression. Previous ex vivo research had failed to establish a link between P-gp function/expression and A61G, G1199A or C1236T SNPs. Research has indicated a lack of influence of G2677T SNP on the ex vivo P-gp expression. However, there is evidence that G2677T SNP does indeed affect the transport capacity by diminishing it [93]. For the C3435T SNP there is some evidence relating to decreased transport and function, but the results have been too varied to achieve any concrete conclusion. In terms of haplotypes, 1236T/2677T/3435T, was found to have no effect on the ex vivo tissues pertaining to expression and function, but they have been associated with decreased mRNA expression in the duodenum [93]. But it is again difficult to conclude anything with the existing knowledge.
As previously indicated the BBB P-gp expression may be just as if not more important than the intestinal P-gp expression for opioids such as methadone. But studying the BBB expression of P-gp is humans is substantially more challenging, but there were two post mortem investigations that indicated 3435C/C genotype may be directly proportional to high P-gp expression in the temporal lobe, though the results were not statistically significant [93]. Thus, we can conclude that despite being inconclusive, the ex vivo studies do strongly indicate that ABCB1 genetic variants can influence the expression and function of P-gp in humans with the best evidence being presented by the experiments dealing with G2677T and C3435T SNPs. But unfortunately, as with a lot of opioid related studies no in vitro or ex vivo studies investigated the impact of ABCB1 genetic variability on opioid transport.
6.7. Functional Effects on Opioids
Studies in healthy subjects investigating the plasma concentrations and respiratory depressive effects or oral loperamide did not find any significant relationship between ABCB1 3435 variant [94]. But the experiment did indicate that the morphine brain distribution was affected by this SNP. Experiments involving pharmacokinetic modelling of morphine in the plasma as well as the CSF of patients undergoing neurosurgery has revealed a significant connotation between homozygous mutant genotype and increased concentration of morphine in the CSF [95] but what is the clinical significance, that is yet to be determined. In another experiment, there was no significant association observed between 3435 SNP and post-operative morphine administration in surgery patients [96]. While testing the requirement of morphine as a pain relief for cancer patients [97], demonstrated that for the first week of the treatment there was a significant association between 3435 SNP and greater relief from pain, within the homozygous wild type (C/C) and homozygous variant (T/T) genotypes predicting 68% of morphine non-responders and 63% of responders respectively. Thus, it was determined that 3435 genotypes were far better at predicting morphine effects than morphine doses which vary between 10-500 mg/day. There was also no correlation between this and the changes in the pain score. This can be concluded as a strong clinical evidence of involvement of ABCB1 genetic variations in early-stage morphine effectiveness for eating cancer pain [98].
As previously mentioned, the ABCB1 gene is highly polymorphic, with significant disequilibrium present in the linkage between variant loci. Hence it was suggested that the effect of ABCB1 haplotypes should be considered instead of individual SNP, they may be able to predict P-gp expression and function more accurately [99]. This may be true in the case of opioids as well. For example, Skerke et al., 2003 indicated no significant association between ABCB1 3435 variant and oral loperamide relating to its intestinal absorption/CNS effects. The investigation into the haplotypes revealed that individuals carrying the ABCB1 haplotype G2677/T3435, had significantly higher, almost 1.5 times higher plasma loperamide concentration as compared to non-carriers. Similarly, Coulbault et al., 2006 found that ABCB1 GG2677/CC3435 diplotype was better at predicting side effects of morphine than the two SNPs analyzed individually [97].
6.8. Other Members of the ABC Family
There are other members of the ABC family of transporters, namely ABCC1 (multidrug resistance-associated protein 1), ABCC2 (multidrug resistance-associated protein 2), and ABCC3(multidrug resistance-associated protein 3). The most likely candidate to be involved in opioid transport among these is the ABCC1, one of the main reasons this could be a possibility is attributed to the level of expression. Ubiquitously expressed ABCC1 will have an advantage over ABCC2 expressed in the brain and ABCC3 expressed in the liver, kidneys and intestine [100]. Direct involvement of ABCC in the transport of opioids is available only for the peptide DPDPE, tested in animal studies. There is indirect evidence that ABCC helps transport morphine [100] as well as other peptides such as endomorphin-1 and 2, met-enkephalins [100]. All three ABCC transport numerous glucuronides [101] hence it could be postulated that they could be involved in the efflux of opioids, but this is yet to be proven. It has two major identified haplotypes G671V variant and a V188E-C1515Y with G671V variant having 28% allelic frequency in the Caucasian population [102]. While the other variant V417I is distributed as follows, Asians 13%–19%, Africans 14%, and Caucasians 22%–26% [102]. Despite having two genetic variants, very few have demonstrated functional consequences related to the transport of opioids. Another member similar to ABCC1 is ABCG2 that was also discovered in the MDR cell line and is also known as BCRP (breast cancer resistance protein) [103]. More than 80 polymorphisms have been found in this gene, among them SNP C421A is widely studied. It is widely distributed among many ethnicities with frequencies such as 27%–35% in Asians, 9%–14% in Caucasians, and 1%–5% in Africans [104]. However, just like with ABCC family, there is no significant link established between ABCG2 and opioid transport.
7. DISCUSSION
It can be observed that the mutations resulting in functional changes in ABCB1 genetic variants are relatively moderate and substrate dependent. While there are functional changes, it cannot be equated as a near loss in function. Nonetheless there is evidence present that suggests clinically significant effects due to polymorphisms at positions 2677 and 3435. The effects being P-gp transport of opioids, particularly morphine, trying to control for the effect of other common ABCB1 genetic variants may also be critical. Genetic variability in the ABCB1 may be important in determining morphine exposure during the administration of illegal opioids, ABCB1 variants are expected to increase opioid exposure during the initial stages or opioid use as well as abuse, which in turn can increase the risk or dependence. There has not been a detailed study about the potential impact of ABCB1 genetic variation on methadone or buprenorphine exposure when used during opioid substitution treatment, as potential P-gp substrates, ABCB1 mutations may result in a reduced dosage requirement and could finally influence the treatment options.
There is a substantial amount of variability among individuals when it comes to opioid response, yet till date this is not taken into consideration when dispensing opioids. This variability stresses the need to individualized treatment strategies that should be aiming for maximizing antinociceptive effect and minimizing dependence/abuse. Unfortunately, the current methods employed as a part of individualizing patient care are only partly successful in retaining those under treatment with a clear majority dropping out. Monitoring plasma drug concentration has been suggested as a potential solution to many of the problems associated with maintenance dosing (Wolff & Strang, 1999). But past experience has shown that target plasma concentrations vary largely between individuals (Dyer & White, 1997). Hence applying the principles of pharmacogenetics, mainly to identify the factors that affect the dose-plasma concentration could prove to be a valuable tool in establishing individualized target doses and plasma concentrations that are required for opioid substitution treatment.
This review article was primarily based around the ABCB1 gene as it is a viable candidate for this type of therapeutic approach. The main reason being its primary product the P-glycoprotein efflux transporter (P-gp). P-gp, as we have seen earlier, can limit the intestinal absorption of the opioids and influences its distribution in the brain. In addition to that the ABCB1 gene is highly polymorphic in nature, and as we have seen before, there are a few genetic variants that may be considered clinically viable to produce the desired effect/changes in an opioid response.
While formulating this article, there are several gaps in the published literature that I noticed and further research is required to fill them up before pharmacogenetics could be considered a useful approach. ABCB1 was the main target for cancer research for years and its role in opioid dependence is slowly starting to catch traction but there needs to be a study observing the actual differences in patient response due to their genetic variability during an opioid substitution treatment. Also, there is no study that defines the relationship between ABCB1 genetic variation and direct susceptibility to opioid addiction and dependence. Also, the direct effects of long-term illicit heroin abuse or administration of methadone or buprenorphine on P-gp expression or its effects on functions in human beings is yet to be examined. The effect of ABCB1 variation on tissue expression and the function of P-gp in opioid substitution is not fully explored. Also, the In vitro transport of methadone and β-endorphins is still unconfirmed in human P-gp expressing cells. There could also be a potential study targeting the question of hereditary, whether the progeny of an opioid user is susceptible to abuse on exposure for the first time?
CONCLUSION
Genomic variation clearly influences sensitivity to pain and can also play an important role in developing chronic pain [5]. As we have seen genetic variation can in fact, influence response to any potential pharmacotherapy, however, genetic factors will be able to provide only a partial picture pertaining to interindividual variability. Other factors such as biological variations (age, gender, ethnicity), environmental factors, co-morbidity and co-medications should also be considered because they together affect the pharmacokinetics as well as pharmacodynamics of potential treatment for pain management. Further research is required to link and characterize the effect of multiple genes along with demographics and clinical variables when trying to individualize the opioid treatment option. Large randomized trials need to be executed to develop optimized algorithms that decide the optimal dosage based on the biological and social information provided by the patient. It is also imperative to continue the development of regulator-approved genotyping assays that will aid clinicians in their routine follow-ups for chronic and acute pain management. Thus, there are a lot of questions to be examined and the ultimate goal in this line of research is to successfully identify the intricate role of ABCB1 genetical variability in opioid response, that may provide clinicians the tool for optimizing the dose leading to a successful treatment.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.