
RESEARCH ARTICLE
Changes in Spinal Cord Following Inflammatory and Neuropathic Pain and the Effectiveness of Resiniferatoxin
Mruvil Abooj, Mahendra Bishnoi*, Christine A. Bosgraaf, Louis S. Premkumar
Article Information

Identifiers and Pagination:
Year: 2016Volume: 9
First Page: 1
Last Page: 14
Publisher ID: TOPAINJ-9-1
DOI: 10.2174/1876386301609010001
Article History:
Received Date: 31/3/2015Revision Received Date: 20/11/2015
Acceptance Date: 24/11/2015
Electronic publication date: 14/3/2016
Collection year: 2016

open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution-Non-Commercial 4.0 International Public License (CC BY-NC 4.0) (https://creativecommons.org/licenses/by-nc/4.0/legalcode), which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
Abstract
Peripheral inflammation or nerve injury results in changes in the spinal cord, initiating a process of central sensitization. Although nociceptive Transient Receptor Potential (TRP) channels have been studied extensively, the role of these channels expressed at the central terminals in the spinal cord is not fully understood. Here, we studied the expression and function of TRPV1 channels at the spinal cord following induction of inflammatory pain by Complete Freund's Adjuvant (CFA) and neuropathic pain by Chronic Constriction Injury (CCI). Rats treated with CFA or subjected to CCI developed long-term thermal and mechanical hypersensitivity. Peripheral inflammation or injury induced an inflammatory response at the levels of spinal cord, which included activation of glia and increased levels of proinflammatory mediators. As a result, expression of TRPV1 was significantly increased and the associated function of TRPV1-mediated CGRP release was also significantly increased. Single intrathecal administration of resiniferatoxin (RTX), an ultrapotent TRPV1 agonist, selectively reversed inflammatory thermal hypersensitivity and the associated changes in TRPV1 expression and function without affecting mechanical hypersensitivity. In summary, peripheral nerve activity triggers an inflammatory response at the spinal dorsal horn, which results in enhanced expression and function of TRPV1 channels. Targeting TRPV1 expressed in the central terminals is a viable strategy to alleviate certain modalities of pain.
INTRODUCTION
Inflammatory and neuropathic pain is a symptom of several pathologies, including but not limited to arthritis, cancer and diabetes. Irrespective of the underlying pathology, the signs and symptoms are often the same: spontaneous pain, hyperalgesia, allodynia and pain summation. Peripheral injury-induced neuronal activity results in an inflammatory response at the spinal cord, which includes release of Calcitonin Gene-Related Peptide (CGRP) and Substance P (SP) from presynaptic nerve terminals and activation of glia influencing the synthesis and release of TRPV1 agonist anandamide and pro and anti-inflammatory mediators. Anandamide and inflammatory mediators act retrogradely to modulate synaptic transmission and TRPV1 expression and function, respectively. Inflammatory response at the spinal cord contributes to central sensitization in the spinal cord, resulting in long lasting thermal hyperalgesia and mechanical allodynia [1-3].
Transient Receptor Potential Vanniloid 1 (TRPV1) receptor, a non-selective cation channel has a high Ca2+ permeability and belongs to a family of TRP channels [2, 4-8]. Mice lacking TRPV1 receptors exhibit decreased sensitivity to capsaicin and thermal stimulus, implying that TRPV1 is essential for certain modalities of pain [5, 9]. TRPV1 receptors are also expressed in the central terminals of sensory neurons in the spinal cord [4]. Activation of TRPV1 promotes Ca2+ influx into nerve terminals, resulting in the release of neuropeptides like CGRP and SP along with glutamate into the synaptic cleft. These neuropeptides activate their respective receptors (CGRP: calcitonin receptor-like receptor and receptor activity-modifying protein 1; SP: neurokinin 1 receptor), which further activate glia and sensitize second order neurons, thus initiating the process of central sensitization [2, 3, 7, 10, 11]. Blockade of CGRP receptors has been effective in treating animal models of inflammatory or neuropathic pain [12-15].
Studies have shown that resiniferatoxin (RTX), an ultrapotent TRPV1 agonist can be an effective long-term analgesic in a number of animal models [6, 16-23]. RTX exhibits these effects by its ability to cause depolarization block of the peripheral or central terminals in the short-term and by nerve terminal ablation in the long-term [24, 25]. There is an ongoing clinical trial to treat terminal debilitating pain conditions using intrathecal RTX administration [26; Clinicaltrials.gov: NCT00804154]
In this study, we have used animal models of inflammatory (CFA-induced) and neuropathic (CCI-induced) pain and determined the changes at the spinal cord that include expression and function of TRPV1. Then, we determined the usefulness of intrathecal administration of RTX in alleviating pain associated with these models.
MATERIALS AND METHODS
Animals
Experiments were conducted on male Sprague Dawley (250-270 g, n=6-8/group) rats purchased from Harlan laboratories, Indianapolis, IN, USA. The animals were housed under standard laboratory conditions, maintained on a normal light-dark cycle and free access to food and water. The experimental procedures minimized pain, distress and unnecessary discomfort and were approved by the Southern Illinois University School of Medicine Institutional Animal Care and Use in accordance with the Guide for the Care and Use of Laboratory Animals.
CFA and RTX Administration
Rats were acclimatized to the test conditions 1 hour/day for 5 days before starting the experiments. After having baseline values for thermal and mechanical testing, animals were divided in two groups. Vehicle treated rats (n=6) received 100 µl of saline in left hind paw and/or intrathecal (i.t.) saline. Each rat in CFA-treated group (n=15) received 100 µl of CFA in left hind paw. Animals were assessed for thermal hyperalgesia and mechanical allodynia 3 and 7 days after vehicle/CFA administration. After the experiment on day 7, CFA-treated animals were divided in 3 groups, i.e. CFA-treated, CFA + i.t. RTX (2 µg/kg/20 µl) and CFA + i.t. RTX (4 µg/kg/20 µl). Each rat in CFA + i.t. RTX (2 µg/kg/20 µl) group received intrathecal (L4/L5 intraspinal space) injection of 2 µg/kg of RTX in 20 μl of saline [23]. Each rat in CFA + i.t. RTX (4 µg/kg/20 µl) group received intrathecal (L4/L5 intraspinal space) injection of 4 µg/kg of RTX in 20 μl of saline. Animals received RTX injections under isoflurane anesthesia (2% oxygen) as described previously [23]. Doses for RTX treatment were selected on the basis of previous studies in our lab. Thermal hyperalgesia and mechanical allodynia was assessed for another 5 weeks. All the experiments involving behavioral studies were performed by the experimenter who was unaware of treatments [22-24].
CCI and RTX Administration
Rats were acclimatized as described above. The rats were anesthetized using intraperitoneal (i.p.) injection of ketamine (85 mg/kg) and xylazine (5 mg/kg). The common sciatic nerve of the left hind paw was exposed at the level of the middle of the thigh by blunt dissection through the biceps femoris. Proximal to the sciatic trifurcation, approximately 7 mm of nerve was freed, and 4 loose ligatures of 4-0 chromic gut were placed around the sciatic nerve. In sham-operated animals were subjected to the same surgical procedure without the ligations. After surgery, all animals were applied analgesic cream containing local anesthetic (Lidocaine) and subcutaneously injected with amoxicillin/clavulinic acid antibiotic. Animals were assessed for thermal hyperalgesia and mechanical allodynia 7 days post-surgery. After the experiment on day 7, CCI animals were divided in 3 groups, i.e. CCI group, CCI group + i.t. saline, CCI + i.t. RTX (2 µg/kg/20 µl) group [27]. Animals received RTX injections under isoflurane anesthesia (2% oxygen).
Measurement of Thermal Sensitivity:
Thermal nociceptive responses were determined using a plantar test instrument (Ugo Basile, Camerio, Italy). The rats were habituated (15 minutes acclimation period) to the apparatus that consisted of three individual Perspex boxes on a glass table. A mobile radiant heat source was located under the table and focused onto the desired paw. Paw Withdrawal Latencies (PWLs) were recorded three times for each hind paw and the average was taken as the baseline value. A timer was automatically activated with the light source, and response latency was defined as the time required for the paw to show an abrupt withdrawal. The apparatus has been calibrated to give a normal PWL of approximately 6-12s. In order to prevent tissue damage a cut-off at 20s was used.
Measurement of Mechanical Sensitivity:
Mechanical nociceptive responses were assessed using a dynamic plantar anesthesiometer instrument using von Frey hairs (Ugo Basile, Camerio, Italy). Each rat was placed in a chamber with a metal mesh floor and was habituated (15 min acclimation period) to the apparatus. A 0.5 mm diameter von Frey probe was applied to the plantar surface of the rat’s hind paw with pressure increasing by 0.05 Newton/s and the pressure at which a paw withdrawal occurred was recorded and this was taken as Paw Withdrawal Threshold (PWT). For each hind paw, the procedure was repeated 3 times and the average pressure to produce withdrawal was calculated. Successive stimuli were applied to alternating paws at 5 min intervals.
CGRP Release Assay:
1) Tissue preparation: Five rats from each group were used. Animals were anesthetized using isoflurane (5%) and oxygen. Spinal cord (hydraulic method) and paw skin was dissected quickly. Lumbar portion of spinal cord (0.10 g, range 0.05–0.15 g) and skin flaps (0.30 g, range 0.25-0.40 g) were collected and were kept for 30 min in SIF (107.8 mM NaCl, 26.2 mM NaCO3, 9.64 mM Na-gluconate, 7.6 mM sucrose, 5.05 mM glucose, 3.48 mM KCl, 1.67 mM NaH2PO4, 1.53 mM CaCl2, 0.69 MgSO4) gassed with 95% oxygen and 5% carbon dioxide at 32°C for 30 min. These segments were then taken for elution studies.
2) Elution procedures and stimulation: A series of 3 glass tubes were filled with 0.5 ml SIF each and positioned in a temperature controlled shaking bath (32°C). The release experiment was started by transferring the mounted spinal cord segment/paw skin flap into the first tube. After 5 min incubation, spinal cord segments/ paw skin flap were transferred to the second tube for another 5 min and then to the third tube for 5 min. After two control samples, stimulation was performed in the third tube with capsaicin (10 ìM, Sigma) prepared from the stock (100 mM in ethanol).
For CGRP release assay, 200 μl of elutes were taken from each tube. Spinal cord elutes were further centrifuged at 2000 rpm for 10 minutes. Samples were mixed immediately after the incubation with 50 μl of 5-fold concentrated commercial CGRP EIA buffer containing several protease inhibitors. The CGRP content was determined immediately after the end of the experiment using commercially available enzyme immunoassays (CGRP: SPlbio, France, detection limit=8-10 pg/ml). All EIA plates were determined photometrically using a microplate reader.
Immunohistochemistry:
Immunofluorescence labeling of TRPV1 receptors and microglial marker (OX-42) and astrocyte marker (GFAP) in spinal dorsal horn was performed on three vehicle- treated, three sham-operated, three CFA-treated and three CCI rats to determine the effect of CFA-treatment and CCI on TRPV1 receptor expression and microglial activation in spinal dorsal horns. Rats were deeply anesthetized with i.p. injection of ketamine (85 mg/kg) and xylazine (5 mg/kg) and perfused intracardially with 100 ml ice-cold normal saline followed by 150 ml 4% paraformaldehyde in 0.01 M PBS (pH 7.4). The lumbar segment of the spinal cord were quickly removed and post-fixed for 2 hr in the same fixative solution and cryoprotected in 20% sucrose in 0.01 M PBS for 24 hr at 4°C. Following cryoprotection, 15 μm sections were cut from lumbar portion of spinal cords using a microtome (Leica CM 1850, North Central Instruments Inc, Plymouth MN, USA).
For TRPV1 receptor, microglial marker (OX-42) and glial fibrillary acidic protein (GFAP) immunostaining, the sections were rinsed in 0.1 M PBS and permeated with 0.1% triton X in 0.01 M PBS for 30 minutes. The sections were then blocked in 10% normal donkey serum in PBS for another 30 minutes. The sections were then incubated overnight with the primary antibody (TRPV1: Guinea anti-TRPV1 N terminal (dilution 1:1000) Neuromics, Minneapolis, MN, USA and microglial activation marker: Mouse anti-CD11b (OX- 42) (dilution 1:100), mouse anti-GFAP antibody (1:100), Novus Biologicals, Littleton, CO, USA) diluted in PBS containing 1% normal donkey serum, 0.1% Triton X-100. Subsequently, sections were rinsed in PBS and incubated (1 hour) with the secondary antibody (Rhodamine donkey anti guinea pig IgG (dilution 1:50), Jackson Immuno-research Laboratories, PA, USA) diluted in PBS containing 1% normal donkey serum, 0.1% Triton X-100. Sections were washed and fixed. A confocal microscope (Specifications: Intensity- 20%, Gain-1 (4 for microglia slides), Zoom-1, Offset-0, Scan speed 150-160 seconds, Filter and color tool-normal) was used to view the sections, and areas of interest were photo documented and quantified.
Western Blotting:
Animals were anesthetized using isoflurane (5%) and then sacrificed by decapitation. Lumbar portion of spinal cord were collected and kept in lysis buffer. Protein levels were determined as described previously [28]. Lysis buffer contains 1X PBS buffer containing 1x Protease Inhibitor (Roche, Indianapolis, IN), EGTA and Phosphatase inhibitors (Phosphatase inhibitor cocktail 1, Sigma and Phosphatase inhibitor cocktail 2, Sigma). Samples were homogenized using sonicator. The tubes were spun at 10,000 rpm for 15 min (at 4oC) to remove cellular debris. Protein concentration in supernatants was determined by using the Pierce BCA Protein Assay Kit (Thermo Scientific) as per the manufacturers’ protocol. Proteins (40 to 80 ìg) were separated by a 12% SDS-PAGE polyacrylamide gel and transferred to a nitrocellulose membrane. The membrane was blocked with 5% non-fat milk (Lab Scientific, inc.) dissolved in 0.05% TBS-T at room temperature for 1h (TBS :Tris Buffered Saline was prepared using the formula: 50 mM Tris-HCl, 150 mM NaOH, pH 7.5. 0.05% TBS-T was prepared by adding 50 ml of Triton-X [MP Biomedicals, LLC] to 100 ml of TBS). The membrane was then rinsed with 0.05% TBS-T and then incubated overnight at 4oC in primary antibody. The primary antibodies used were TRPV1 rabbit polyclonal antibody (Santa Cruz) and Tubulin Mouse antibody (1:5000) (Santa Cruz) used as a loading control. The primary antibody solution was removed and the membrane was washed with 0.05% TBS-T, 3 times for 10 min each. The membrane was treated using anti-rabbit antibody (IR800) (1:10,000) or anti-mouse antibody (IR700) (1:5000) for 1 hr at room temperature. The secondary antibody solution was removed and the membranes were washed with 0.05% TBS-T, 3 times for 10 min each. All the antibody dilutions were made in SuperBlock Blocking Buffer in TBS (Thermo Scientific). The blots were analyzed using the Li-Cor Odyssey infrared imaging and densitometric analysis was performed using the Li-Cor Odyssey software.
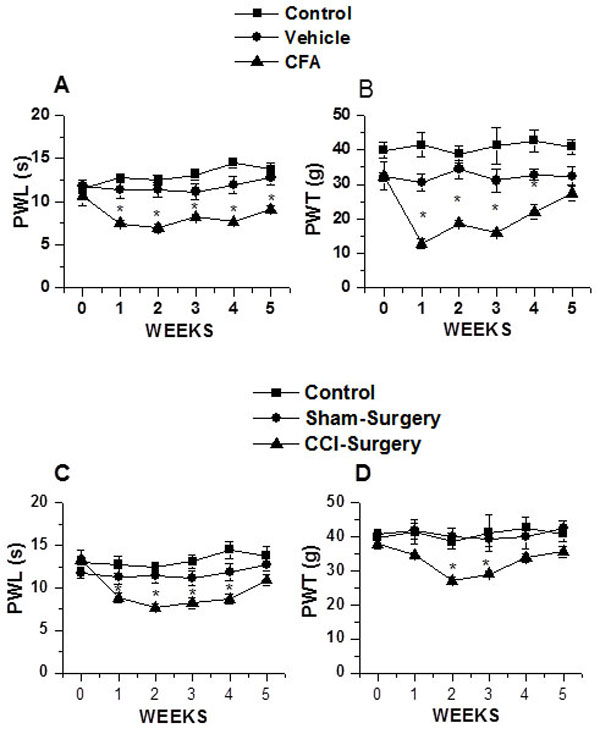 |
Fig. (1). Effect of intraplantar CFA and CCI on thermal hyperalgesia and mechanical allodynia. A. Intraplantar CFA administration resulted in significant (p<0.05) decrease in PWL at week 1 and lasted for entire duration of study compared to vehicle-treated group. B. Intraplantar CFA administration resulted in significant (p<0.05) decrease in PWT at week 1 after treatment compared to vehicle-treated group thus inducing thermal hyperalgesia and mechanical allodynia. C. CCI resulted in significant (p<0.05) decrease in PWL at week 1 and returned to baseline by week 5 of study compared to vehicle-treated group. D. CCI resulted in significant (p<0.05) decrease in PWT at week 2 and returned to baseline by week 4 of study compared to vehicle-treated group. Asterisks (*) represent p < 0.05 as compared to vehicle-treated or sham-surgery animals. |
Statistical Analyses:
Statistical analysis was performed using SPSS software. Data are presented as mean ± SEM. Student T test was used to compare two groups. Descriptive one way ANOVA followed by post hoc Tukey’s HSD was used to compare more than 2 groups. A p value of <0.05 was considered as statistically significant.
RESULTS
Effect of intraplantar administration of CFA and CCI of the sciatic nerve on thermal hyperalgesia and mechanical allodynia
Intraplantar administration of CFA and CCI by sciatic nerve ligation were used as models of peripheral nerve injury-induced pain. First, we characterized the pain thresholds for each of these two pain models. Pain thresholds were measured by the two commonly used techniques: 1) Thermal pain testing (Hargreaves’ test) and 2) Mechanical pain testing (von Frey test). Animals were acclimatized in plexi-glass chambers for a week before testing for thermal and mechanical pain sensitivities. Animals were randomly divided into four groups: 1) vehicle-treated; injected with intraplantar saline, 2) CFA-treated; injected with intraplantar 100 µl CFA, 3) sham-operated and 4) CCI-induced by sciatic nerve ligation.
 |
Fig. (2). Altered OX-42 (Microglial activation) and GFAP (astroglial activation) staining in spinal cord (L4-L5) dorsal horn of CFA-treated and CCI-induced rats. A,B,C. Representative images of OX-42 staining from a vehicle-treated, CFA-treated and CCI rats. D,E,F. Representative images of GFAP staining from a vehicle-treated, CFA- treated and CCI rats. [Scale bar is 50 μm]. |
Intraplantar administration of CFA significantly decreased PWL (vehicle: week 0, 11.58±0.76s, week 1, 12.75±0.23s, n=6; CFA: week 0, 10.69± 1.1s, week 1, 7.48± 0.36s, n=12, p<0.05) (Fig. 1A). Thermal hyperalgesia lasted beyond 5 weeks (vehicle: week 5, 13.7± 0.69s; CFA: week 5, 9.11± 0.42s, p<0.05) (Fig. 1A). PWT also was significantly decreased following CFA administration (vehicle: week 0, 32.29± 1.25g, week 1, 30.61± 2.34g; n=6; CFA: week 0, 32.42± 3.98g, week 1, 12.92± 1.54g, n=12, p<0.05) (Fig. 1B). Mechanical allodynia lasted for 4 weeks and there was no difference in PWT between vehicle-treated and CFA-treated animals by the end of week 5 (vehicle: week 5, 32.42± 2.54g; and CFA: week 5, 27.41± 2.21g) (Fig. 1B).
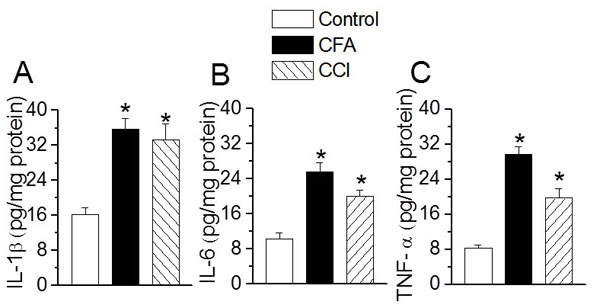 |
Fig. (3). Effect of intraplantar CFA and CCl on spinal pro-inflammatory cytokine levels. CFA-treatment and CCI resulted in a significant increase in interleukin-1beta (IL-1β) (A) Interleukin-6 (IL-6) (B) Tumor necrosis factor-α (TNF-α) (C) In lumbar spinal cord tissue homogenates as assessed by ELISA. Statistically significant data are denoted by an asterisk (*), p<0.05 as compared to control or vehicle-treted rats. |
Chronic constriction nerve injury significantly decreased PWL (sham-operated: week 0, 11.78± 0.65s, week 1, 11.32± 0.96s; CCI-induced: week 0, 13.32± 1.14s, week 1, 8.85± 0.65s, p<0.05) (Fig. 1C). In these experimental conditions, the thermal hyperalgesia lasted for 4 weeks following CCI and the values were not significantly different by week 5 (sham-operated: week 5, 12.78± 1.02s; and CCI: week 5, 10.88± 0.66s; Fig. 1C). The PWT also significantly decreased after 2 week (sham-operated: week 0, 40.90± 1.25g; week 3, 40.06± 2.58g; CCI-induced, week 0, 37.86± 2.46g; week 3, 27.13± 1.58g, p<0.05) (Fig. 1D). This decrease in PWT following CCI lasted for 3 weeks and was not significantly different from sham-operated by week 5 (sham-operated: week 5, 42.77±1.89s; CCI-induced: week 5, 35.63±1.65s) (Fig. 1D). These data suggest that there are differences in the extent of nerve injury, which can be correlated with the degree of thermal hyperalgesia and mechanical allodynia caused by these two different models of peripheral nerve injury. Intraplantar CFA administration has a long lasting effect on thermal hyperalgesia as compared to the CCI.
Microglial and Astroglial Activation Following Intraplantar CFA Administration and CCI
Microglial and astroglial activation plays a key role in maintenance of thermal hyperalgesia and mechanical allodynia. Here we determined the glial activation by immuno histochemical staining of spinal cord sections. We used OX-42 staining for demonstrating microglial activation, while GFAP staining was used to visualize activated astrocytes. Following either CFA-treatment or CCI, there was a qualitative increase in OX-42 staining (Figs. 2A, B, C) as well as GFAP staining (Figs. 2D, E, F) in laminae I and II. These data suggest that in both the models of peripheral injury caused activation of microglia and astroglia in the spinal cord.
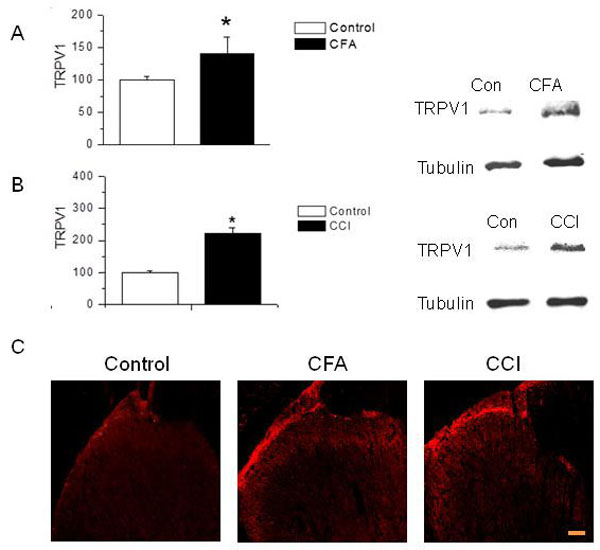 |
Fig. (4). Expression of TRP channels in CFA-treated and CCI-induced animals. A. Intraplantar CFA administration resulted in significant (p<0.05) increase in expression of TRPV1 in spinal cord as compared to vehicle treated group. B. CCI resulted in significant (p<0.05) increase in expression of TRPV1 in spinal cord tissue as compared to vehicle-treated group. Asterisks (*) represent p<0.05 as compared to vehicle-treated animals. C. Immunohistochemical staining shows enhanced expression of TRPV1 in spinal dorsal horn in CFA-treated and CCI-induced animals. |
Effect of Intraplantar CFA and CCI on Spinal Pro-inflammatory Cytokines
Recent literature has provided substantial evidence for the role of pro-inflammatory mediators in the initiation and maintenance of thermal hyperalgesia and mechanical allodynia (1). CFA-treatment and CCI significantly increased the levels of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α) in the spinal cord homogenates (IL-1 β: vehicle, 16.32± 1.55; CFA, 35.68±2.56 ; CCI, 33.26±365 pg/mg protein, n = 5, p < 0.05, Fig. (3A) ; IL-6: vehicle, 10.25± 0.65; CFA, 25.69± 1.86 ; CCI, 19.69± 2.25 pg/mg protein, n = 5, p < 0.05, Fig. (3B) ; TNF-α: vehicle, 8.32± 0.65; CFA, 29.65± 1.86 ; CCI, 19.69± 2.25 pg/mg protein, n=5, p < 0.05, Fig. (3C). These data suggest that the increases in pro-inflammatory mediators in the spinal cord are associated with peripheral nerve activity in CFA- and CCI-induced pain.
TRPV1 Expression Following Intraplantar Administration of CFA and CCI
Next, we wanted to compare the changes in TRPV1 expression at the height (week 2) of thermal hyperalgesia and mechanical allodynia. We accomplished this goal by measuring changes in the expression of nociceptive ion channels TRPV1 at the L4-L6 segments of spinal cord. The Western blot analysis of spinal cord samples at week 2 showed that both, CFA-treated and CCI-induced animals revealed a significant increase in expression of TRPV1 (CFA-treated, 1.5 fold and CCI-induced, 2 fold, respectively; n=3; p<0.05, Figs. (4A and 4B). We also performed immuno-histochemical analysis to visualize TRPV1 expression in spinal cord. Both CFA-treatment and CCI resulted in an increase in intensity of TRPV1 in lamina I of dorsal horn of spinal cord compared to vehicle-treated group (Fig. 4C). These results suggest a significant increase in the expression of TRPV1 in the central sensory nerve terminals that form synapses with the second order neurons. TRPV1 has been shown to be expressed only in sensory nerve terminals in the spinal cord [24, 29, 30]. However, recently it has been shown that TRPV1 is also expressed in inhibitory interneurons in the spinal cord [31].
Intraplantar CFA Administration and CCI on TRPV1-Mediated CGRP Release
As indicated earlier, both peripheral and central terminals of the sensory neurons express TRPV1. Following the observation that intraplantar CFA administration and CCI increased the expression of TRPV1 in spinal dorsal horn, we hypothesized that intraplantar CFA administration and CCI affect TRPV1-mediated functions such as neuropeptide release in central terminals of DRG neurons [22-24]. TRPV1-mediated CGRP release is a reliable assay to determine the expression and function of TRPV1. The neuropeptide release was normalized to the weight of the spinal cord tissue. The basal CGRP levels were significantly increased in both CFA-treated and CCI-induced groups as compared to vehicle-treated group at week 2 (vehicle, 12.74± 0.9; CFA, 18.5± 1.71; CCI, 21.56± 0.99 ng/g tissue/5min; p<0.05) (Fig. 5A). To specifically compare the role of TRPV1, we examined capsaicin-evoked CGRP release. Incubation of spinal cord tissue with capsaicin (1 µm) for 5 min significantly increased CGRP release in vehicle-treated and CFA-treated animals. The magnitude of 1 μM capsaicin-stimulated CGRP release was significantly greater in CFA-treated and CCI-induced rats at week 2 (vehicle, 42.56± 4.22; CFA, 58.55± 5.5; CCI, 71.45± 9.6 ng/g tissue/5min; p<0.05) (Fig. 5B). These data demonstrate that TRPV1 expression and functions are enhanced in the spinal cord following peripheral administation of CFA or following CCI of sciatic nerve.
 |
Fig. (5). Change in basal and capsaicin-evoked CGRP release after intraplantar CFA administration and CCI. A. Both intraplantar CFA administration and CCI resulted in significant (p<0.05) increases in the basal-CGRP release as compared to vehicle treated group. B. Both intraplantar CFA administration and CCI resulted in significant (p<0.05) increase in the capsaicin evoked-CGRP release as compared to capsaicin evoked-CGRP release in vehicle treated group. Capsaicin evoked-CGRP release in vehicle-treated group was significantly higher than basal CGRP release in vehicle treated group. Statistically significant results are denoted by asterisks (*) and (**) represent p<0.05 or p<0.01 as compared to basal-CGRP and capsaicin evoked-CGRP release in vehicle-treated rats, respectively. |
Effect of Intrathecal Administration of RTX on CFA- and CCI-Induced Thermal Hyperalgesia and Mechanical Allodynia
In this section, we determined whether intrathecal administration of RTX can alleviate thermal hyperalgesia and mechanical allodynia in models of peripheral injury that lead to chronic pain conditions. One week after intraplantar administration of CFA, animals were injected with intrathecal RTX (2 and 4 µg/kg/20 µl). We have previously shown that intrathecal injection of vehicle did not cause changes in thermal and mechanical sensitivities [23, 24]. Both 2 and 4 µg/kg dose of RTX significantly attenuated the increase in PWL over the period of 5 weeks (week 1 after RTX injection, vehicle, 13.74± 1.22s; CFA, 9.11± 0.74.s, CFA + RTX (2 µg/kg/20µl), 14.78± 1.05.s; CFA + RTX (4 µg/kg/20 µl), 15.91± 0.96s; p<0.05, Fig. (6A) but didn’t attenuate PWT week 1 after RTX injection, vehicle, 34.33± 2.42g; CFA, 18.72± 1.58g, CFA + RTX (2 µg/kg/20µl), 21.83± 2.62g; CFA + RTX (4 µg/kg/20µl), 19.17± 1.06g; p<0.05, Fig. (6B). There was no significant difference in the efficacy of the two doses, both exhibiting similar profile. Based on these experiments, we suggest that, 2 µg/kg dose of RTX is sufficient to ablate TRPV1 terminals in lumbar region of spinal cord and alleviate thermal hyperalgesia.
 |
Fig. (6). Effect of intrathecal administration of RTX on intraplantar CFA and chronic constriction nerve injury induced thermal hyperalgesia and mechanical allodynia. A,C. RTX attenuated thermal hyperalgesia in both CFA-treated and CCI rats. B,D. RTX administration did not have any effect on mechanical allodynia in both CFA-treated and CCI-induced rats. Statistically significant results are denoted by an asterisk (*), p<0.05. |
Similarly, following one week after surgery to induce CCI, one group of animals was injected with intrathecal RTX (2µg/kg/20µl). Intrathecal administration of RTX significantly attenuated the increase in PWL (week 1 after RTX injection: Sham surgery, 11.78± 0.93s; CCI, 8.88± 1.37s, CCI + RTX (2 µg/kg/20µl), 11.90± 0.85s; p<0.05, Fig. (6C) but did not change PWT (week 1 after RTX injection: Sham surgery, 40.06± 2.35g; CCI, 27.13± 1.74g, CCI + RTX (2 µg/kg/20 µl), 30.47± 2.05g; p<0.05, Fig. (6D). As there is convincing literature suggesting the involvement of TRPV1 in thermal hyperalgesia but not in mechanical allodynia, these findings attest to a selective effect of RTX against thermal hyperalgesia.
Effect of Intrathecal Administration of RTX on TRPV1 Mediated CGRP Release
RTX administration can selectively ablate TRPV1 expressing nerve terminals in spinal cord [24]. In order to confirm that intrathecal RTX administration ablated TRPV1 receptor expressing nerve terminals in the spinal cord, we determined the functionality of TRPV1 receptors using CGRP release assay. RTX treatment (2 µg/kg/20µl) significantly inhibited the increase in capsaicin-stimulated CGRP release in spinal cord samples following CFA-treatment and CCI (vehicle, 42.56± 4.22; CFA, 9.44± 0.87; CCI, 18.26± 1.52 pg/g tissue/5min Fig. (7A). These results suggest that capsaicin-stimulated CGRP release in spinal cord is a TRPV1-dependent process. Intrathecal administration of RTX ablates TRPV1 expressing nerve terminals in spinal dorsal horn; thereby preventing TRPV1-mediated CGRP release.
TRPV1-mediated peripheral CGRP release plays an important role in maintaining vasodilation and regulating micro vascular circulation. In order to confirm that intrathecal RTX can selectively ablate TRPV1 expressing central terminals in the spinal cord, we determined CGRP release from paw skin samples. Treatment with intrathecal RTX (2 µg/kg/20µl) did not significantly change capsaicin-stimulated CGRP release in both CFA-treated (48.81± 9.25 vs. 52.72± 3.86 pg/g tissue/5min) and CCI-induced (84.44± 11.61 vs. 77.81± 5.68 pg/g tissue/5 min; Fig. (7B) animals. These observations suggest that intrathecal injection of RTX does not affect the TRPV1-mediated CGRP release from paw skin, thus preserving peripheral nerve function of CGRP release.
DISCUSSION
Central sensitization has been extensively studied [3] and has been shown to involve glutamatergic, gabaeric and opioidergic systems along with microglial activation, synthesis and release of neuropeptides (CGRP/SP), cytokines and chemokines [1-3, 32-35]. Inhibition of glial activation leads to attenuation of neuropathic pain in several models [36, 37]. Activation of TRP channels expressed in the nerve terminals can lead to influx of Ca2+ and cause glutamate and CGRP release, which can subsequently activate their respective receptors in glia and mediate an inflammatory response [7, 8, 24, 33, 38-40]. Although there is restricted expression of TRPV1 in the central nervous system [24, 29, 30], studies have also shown that TRPV1 is expressed in glial cells [7, 38]. Several pro-inflammatory mediators are released from activated microglia. We have observed that there is a significant increase in the levels of pro-inflammatory mediators IL-1β, IL-6 and TNF-α. These mediators can interact with TRPV1 expressing cells and alter TRPV1 expression and function [41, 42]. Peripheral inflammation (intraplantar CFA-treatment) and peripheral nerve injury (CCI) have been shown to increase the levels of phosphorylated p38 and ERK in spinal cord tissue [43-49]. Activation of MAPKs in glial cells is necessary for the development and maintenance of neuropathic pain [50-52].
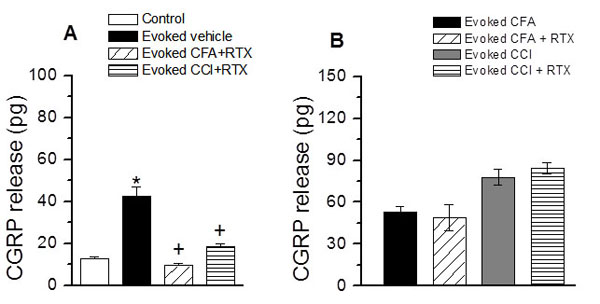 |
Fig. (7). Effect of intrathecal administration of RTX on TRPV1-mediated CGRP release. A. Intrathecal RTX treatment resulted in significant decrease in capsaicin evoked-CGRP release in CFA-treated and CCI-induced rats in spinal cord samples. B. Intrathecal RTX treatment did not significantly change the capsaicin evoked-CGRP release in CFA-treated and CCI-induced paw skin samples. Statistically significant results are denoted by an asterisk (*, increase) and a plus (+, decrease), p<0.05. |
The physiological role of TRPV1 expressed in the central terminals of sensory neurons is not fully understood. Activation of TRPV1 in the central terminals modulates synaptic transmission [24, 33, 53-55]. In order to attribute a role for TRPV1, we determined the expression levels of TRPV1 by Western blot and immunohistochemistry techniques. The Western blot analysis revealed that in both models, there was a significant increase in TRPV1 expression at the dorsal horn. This was further confirmed by immuno-histochemical analysis, which showed an increase in intensity of TRPV1 staining in spinal dorsal horn. In order to confirm that increased TRPV1 expression results in functional changes, we determined TRPV1-mediated CGRP release. CGRP is stored in vesicles of both peripheral and central terminals and is released when extracellular Ca2+ enters the terminals. Enhanced CGRP release occurs from central terminals during peripheral nerve injury and peripheral inflammation [56-58]. There are studies showing that CGRP is involved in peripheral, spinal, and supra-spinal pain mechanisms [57, 59, 60]. The released CGRP binds to its receptor on second order neurons and glia, leading to change in receptor expression and function. Activated glia release inflammatory mediators, which act on their respective receptors in the neurons and alter neuronal activity, thus contributing to central sensitization. Our studies show that in both models, intraplantar CFA-treatment and CCI not only increased basal CGRP release, but also increased TRPV1-mediated CGRP release. These findings further support the hypothesis that peripheral nerve activity leads to increased TRPV1 expression and function in the spinal cord.
The results obtained from this study show that intrathecal (L4-L5) administration of RTX resulted in reversal of PWL induced by intraplantar administation of CFA and CCI of the sciatic nerve; while intrathecal RTX treatment did not show any effect on mechanical allodynia (PWT). This result is consistent with the fact that TRPV1 is major contributor of inflammatory thermal hyperalgesia. Previous studies from our lab and other labs have shown that RTX selectively ablated central terminals of TRPV1 expressing neurons resulting in reversal of thermal hyperalgesia [16, 18-20, 22-24]. We propose that selective ablation/blockade of TRPV1-expressing central terminals is sufficient to cause long lasting pain relief sparing the DRG cell bodies and peripheral terminals intact. We have also reported that intrathecal administration of RTX (targeting central terminals) selectively abolishes inflammatory thermal hypersensitivity without affecting acute thermal pain sensitivity, whereas intraperitoneal administration of RTX (targeting the whole TRPV1 expressing neuron) affects both acute and inflammatory thermal hyperalgesia [23]. Analgesic effects of localized application of RTX can be explained by its ability to cause depolarization block of the peripheral or central nerve terminals in the short-term and nerve terminal ablation in the long term [24, 25, 33]. These observations further validate the claim that TRPV1 is involved in inflammatory thermal but not mechanical hypersensitivity [5, 21, 30, 61], although some studies have shown that selective agonists and antagonists of TRPV1 affected mechanical sensitivity [62-64]. It is possible that TRPV1 expressed in the inhibitory interneurons [31], when blocked reduces inhibition and exaggerates transmission of mechanical sensitivity. While using agonists, it is important to consider the concentrations of agonists used, which may either result in partial or total ablation of TRPV1 expressing nerve terminals impacting the mechanical sensitivity.
Here, we interpret that attenuation of thermal hyperalgesia and CGRP release from central terminals by RTX as ablation of TRPV1 expressing nerve terminals as a result of sustained activation resulting in excessive Ca2+ influx [6, 17, 24]. However, we cannot rule out the possibility that sustained activation of TRPV1 leading to receptor internalization. If only TRPV1 internalization occurs after intrathecal RTX administration and the nerve terminal integrity is maintained, the propagation of action potential should not be impacted. However, we are able to observe a selective alleviation of inflammatory thermal hypersensitivity, which argues against receptor internalization. We also measured CGRP release from peripheral terminals to determine the effect of intrathecal administration of RTX on peripheral TRPV1 function, which show that TRPV1-dependent CGRP release was unaffected following CFA-treatment and CCI. Peripheral TRPV1 function is necessary to maintain micro vascular circulation [2, 65]. TRPV1-mediated CGRP release has been shown to be important for maintaining cardiovascular function [66]. Intrathecal administration of RTX, only affected the central terminals, sparing the rest of the neuron, which included the cell bodies [22, 24]. However, following intraperitoneal administation of RTX, CGRP release from the peripheral and central terminals was abolished as a result of whole TRPV1- expressing neuronal loss [23].
CONCLUSION
Peripheral neuronal activity induced by administation of CFA or CCI causes neuropeptide (CGRP) release. CGRP acting on its receptors triggers glial activation, which alters the extracellular milieu at the dorsal horn by synthesizing and releasing inflammatory mediators. Inflammatory mediators acting on their respective receptors trigger downstream signaling cascades leading to transcriptional, translational and posttranslational changes of receptors, including TRP channels. Targeting central TRPV1 expressed at the dorsal horn by intrathecal administration of RTX, selectively alleviates inflammatory thermal hypersensitivity in a long-term basis, which can be a useful strategy to treat certain modalities of chronic pain.
LIST OF ABBREVIATIONS
- CCI: Chronic Constriction Injury
- CFA: Complete Freund’s adjuvant
- CGRP: Calcitonin Gene Related peptide
- DRG: Dorsal Root Ganglion
- ERK: Extracellular Signal-regulated Protein Kinase
- IL-6: Interleukin6
- IL-1β: Interleukin1β
- MAPK: Mitogen Activated Protein Kinase
- PWL: Paw Withdrawal Latency
- PWT: Paw Withdrawal Threshold
- RTX: Resiniferatoxin
- TNF-α: Tumor Necrosis Factor α
- TRPV1: Transient Receptor Potential Vanilloid 1
- TRPA1: Transient Receptor Potential Ankyrin 1
AUTHORS' CONTRIBUTIONS
MA performed the Western blot, ELISA, designed the study and drafted the manuscript. MB performed the drug administration, surgery, ELISA, immunofluorescence and designed the study. CB performed the behavioral tests. LP designed the study and drafted the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Supported by grants from National Institutes of Health (DA028017) and EAM award from SIUSOM.




